Synopsys and Ansys power the future of innovation—connecting silicon to systems.
ANSYS BLOG
February 8, 2021
MDR and Materials: How Medical Device Manufacturers Can Meet New Regulation Challenges
The European Union Medical Device Regulation of 2017 will take effect beginning May 26. It replaces the EU’s Medical Devices Directive (MDD), and covers all new medical devices produced in and supplied to the EU. In short, medical device manufacturers are facing some of the most disruptive regulations in recent memory. They are turning to technology for help.

One of the biggest changes from MDD to MDR is section 10.4, which restricts the use of substances classified as Category I Carcinogenic, Mutagenic or Toxic for Reproduction (CMRs) and Endocrine Disrupting Chemicals (EDCs) unless a justification is accepted. This new challenge is forcing enterprises to adopt new ways to manage their material information and understand their restricted substances.
Let’s look at some best practices for managing materials information and the key benefits of using a centralized system to manage materials information when addressing section 10.4.1 of MDR.
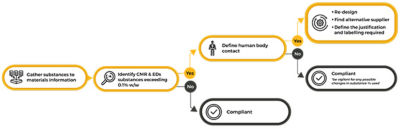
Initial steps of addressing MDR compliance
Section 10.4.1 of MDR establishes that CMRs Category I and EDCs cannot be present in more than 0.1% weight by weight (w/w) when in direct contact with patients or in contact with solids, fluids or gases that are administered or re-administered to patients. Nevertheless, if a manufacturer can show that the use of the substance is justified from a benefit-risk to patient analysis, this restriction can be lifted. To better understand what is in medical devices, substances and materials information needs to be gathered from suppliers and managed. Establishing a companywide approach to manage material and substances information can minimize risk and accelerate the design and approval process of medical devices.
Materials and specification information used in manufacturing is naturally complex due to its wide range of properties and linked networks. Reliable quantification of restricted substances can only be achieved by the correct mapping and tracing of every link in these networks. Further complicating matters is the fact that multiple substances can be used during the manufacturing process that are not part of the final product. Although it is important to track and trace their use, such substances will have no impact on the final MDR approval. Moreover, it is also important to consider the human body contact relevant for MDR when identifying and reporting on substances information for devices approval.

Centralize to Comply
MDR is a new challenge for many medical device companies, but the same information that is being gathered for this regulation can also be used for multiple regulations and legislations around the world, such as the EU’s Registration, Evaluation, Authorisation and Restriction of Chemicals (REACH) or California Proposition 65. Once all materials, substances, specifications, products and the legislations of interest are combined in a central location, compliance reporting becomes an interdependent data problem that is well-suited to a relational database model.

The correct capture and management of materials information is critical to identify restricted substances accurately. To save unnecessary costs resulting from a late identification of a non-compliant material, it is important to design for compliance from an initial concept phase. We will be exploring this topic further in a later blogpost.
In manufacturing companies, multiple systems are used during the design of a new product, such as computer-aided design and engineering (CAD and CAE) or product lifecycle management (PLM). For organizations using multiple materials and producing various products, a commercial-off-the-shelf materials database solution like Ansys Granta MI is a powerful and cost-effective solution to deal with materials information and compliance analytics. Granta MI uses updated data on changing legislation and integrates with all of the systems required during product development.
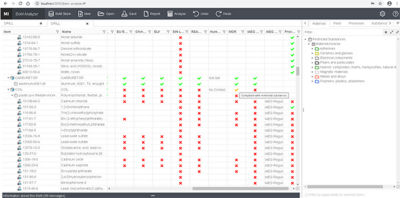
Using a BoM (bill of material) analyzer to cross-reference regulatory compliance of a product in Granta MI
Some of the key benefits of Granta MI include:
- Use of up-to-date reference data on the changing legislation and enable fast compliance assessment for your new and existing products as regulations change.
- Filling in the knowledge gap with reference materials and substance data to assess risk and determine where supplier data is missing.
- Centralizing your materials and substances information in one system to be accessed by multiple teams and departments, accelerating product development and reducing product time-to-market.
- Integrating your material data management system with design tools to ensure consistency and traceability.
- Considering compliance during design, where implementing changes costs least and delivers the most impact.