Synopsys and Ansys power the future of innovation—connecting silicon to systems.
-
-
学生向け無料ソフトウェアにアクセス
Ansysは次世代の技術者を支援します
学生は、世界クラスのシミュレーションソフトウェアに無料でアクセスできます。
-
今すぐAnsysに接続!
未来をデザインする
Ansysに接続して、シミュレーションが次のブレークスルーにどのように貢献できるかを確認してください。
無料トライアル
製品およびサービス
リソースとトレーニング
当社について
Back
製品およびサービス
Ansysブログ
February 20, 2019
医療機器のin silico試験のための規制承認経路の概要
医療機器メーカーはこれまで、ベンチテスト、動物実験、臨床試験(すなわちヒト試験)を実施して、医療機器の安全性と有効性を確立してきました。
これは、機器設計のさまざまな側面に関する意思決定を行う内部プロセスにも、規制当局への承認申請を行う外部プロセスにも当てはまります。
これらの試験には費用と時間がかかる場合があります。さらに、動物実験によってヒトに対する治療の安全性と有効性を予測できるかについて、疑問が呈されています。
in silico評価のための規制承認経路の導入により、ヘルスケア業界でシミュレーションがますます利用されるようになると見込まれる。
医療機器の設計に計算モデリング(すなわちシミュレーションまたはin silico試験)が利用されることが増えています。これらのシミュレーションは、イノベーションを加速させ、長期的な安全性に関する包括的なエビデンスを提供します。しかし、シミュレーションにメリットがあるにもかかわらず、規制当局への申請時に計算モデリングをエビデンスとして活用する動きはあまり見られません。
医療機器のin silico試験のための規制承認経路の導入により、ヘルスケア業界の規制面でシミュレーションがますます利用されるようになると見込まれます。
つい最近まで、「未来の話」だと思われていた医療機器のin silico試験
Fluent社(現Ansys, part of Synopsys)で生物医学エンジニアとして勤めていた私は、ヘルスケア分野においてモデリングの活用と規制への組み込みとの間にギャップがあることを実感しました。
それは2001年、大学院を卒業したばかりの頃です。当時私は、米国食品医薬品局(FDA)医療機器・放射線保健センター(CDRH)の科学技術研究所(OSEL)を含む、医療機器業界のさまざまなクライアントで数値流体力学(CFD)ユーザーをサポートしていました。
規制当局への申請にin silico試験を用いることは一般に「未来の話」と見なされていた。
モデリングは一般に研究ツールとして捉えられ、規制当局への申請資料の一部となり得る信頼性の高い情報源と見なされることがあまりないということは、当初から極めて明白でした。
実際、モデリングを他の医療機器評価手法と同等と見なすことは、未来的、いやむしろ未来主義的な考え方でした。
医療機器のin silico試験の利点
2011年になると、CDRHが計算モデリングを規制科学における優先事項として位置づけました。これにより、in silico試験によって医療機器の性能をより深く理解できることが正式に認められました。
実際、ほかの医療機器評価手法に比べ、in silico研究には際立った魅力的な利点が存在します。利点としては、機器の性能のパラメトリック解析、統計的解析、患者固有の解析を高度に制御された方法で実施できる点などが挙げられます。

in silico試験により、エッジケースをテストできるようになるため、医療機器を評価する際には、すべての患者の健康を考慮することができる。
シミュレーションにより、エッジケースさえも作成し、検討できるようになり、医療機器が最も過酷な状況下で動作できるかについて一定の理解を得ることができます。これは、生理学的な限界領域において統計的に有意な結果を得ることが困難な動物実験やヒト臨床試験とは極めて対照的です。
シミュレーション分野はここ数年にわたり、コンピュータモデリングの規制科学において著しい進歩を遂げてきました。また、FDAなどの規制当局におけるモデリングの活用に関連する重大な障壁も解消されています。これらの障壁は、モデル検証とモデル報告に関連したものです。
FDAが、計算モデリングおよびシミュレーション報告書の作成方法を解説するガイダンスを発表
シミュレーションの規制適用を確立する上で最初に直面する障壁は、計算モデルの枠組みと結果を審査用に要約するための確立された構造がなかったことです。
この課題に対処するため、FDAは計算モデリングおよびシミュレーション(CM&S)報告書の形式と内容を解説するガイダンスを発表しました。
このガイダンスの主な目的は、規制上の意思決定において有効な科学的エビデンスとして機能するだけの十分な信頼性をモデルが有しているかどうかを判断するために必要なすべての情報を申請者が確実に記載できるようにすることです。
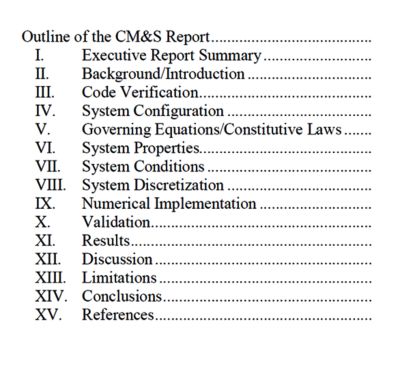
FDAガイダンスに基づくCM&S報告書のアウトライン
また、この文書では、医療機器業界と規制当局が計算モデル研究のさまざまな要素の計画、議論、および審査の過程で使用する共通言語も提供しています。
このガイダンスの本文では、ほとんどの物理学分野に適用できる一般的な報告書形式について説明しています。利用者の便宜を図るため、ガイダンスには、CFD、有限要素法解析(FEA)、電磁界(EM)、超音波、熱伝達、光学に関する詳細な報告推奨事項をまとめた一連の付録もついています。
たとえば、数値電磁界に関する付録では、利用者が用いた支配方程式(Maxwellの方程式など)の概要と、モデル化する医療機器と組織の電気的特性に関する情報を記載することを推奨しています。
医療機器のin silico試験のリスク評価
規制当局への申請におけるシミュレーション結果の利用が限定的であったもう1つの理由は、計算モデルの十分な信頼性を確立するために必要な証拠基準について合意形成が必要であったことです。この重要な課題に対応するために2011年に設立されたのが、米国機械学会(ASME)の「医療機器の計算モデルに関する検証・妥当性確認(V&V)小委員会(ASME V&V 40小委員会)」です。
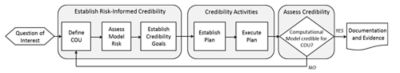
ASME V&V 40規格で定義された、リスクに基づく信頼性評価フレームワーク
医療機器業界、業界サービスプロバイダー、ソフトウェア開発者、およびFDAのメンバーで構成されるこのグループは、リスクに基づく信頼性評価フレームワークを開発しました。
このフレームワークの主要な原則は、コンピュータモデルに基づいてリスクの高い意思決定を行うには、リスクの低い意思決定を行う場合よりも多くの妥当性確認を行う必要があるというものです。
このため、このASME規格では、モデルの信頼性を確立するために必要なV&Vの程度をユーザーがどのように決定すべきかを指針として示しています。この規格は、FEA、CFD、熱伝達に関するASME V&V規格を補完するものです。
in silico医療機器試験の妥当性を確認する方法
7年にわたる努力の末、医療機器のV&Vに関するASME規格が2018年11月19日に発行されました。この規格により、計算モデリングのための規制承認経路が確立されました。
ASME V&V 40小委員会は現在、コミュニティに対し、この規格とそのベストプラクティスに関する啓発活動に取り組んでいます。この活動は、インダストリーデー、トレーニングセッション、事例紹介、および新規作業項目を通じて行われています。
医療機器に関するASME V&V 40小委員会の副委員長として、このように献身的で優秀な科学者および技術者の方々と共に活動できたことは、私にとって名誉であり、大きな喜びです。

V&V 40小委員会は、産業界が社内の意思決定や規制当局への申請を行うために医療機器の信頼性の高いシミュレーションを実施できるよう、in silico試験のベストプラクティスを指導したいと考えている。
Ansysの医療分野への支援についてはin silico試験に関するページ、または業界別ページ『ヘルスケア:医療機器』をご覧ください。
ASME V&V 40小委員会への参加方法については、同委員会の連絡先ページをご覧ください。