Synopsys and Ansys power the future of innovation—connecting silicon to systems.

Why Hydrogen Transport Remains a Critical Engineering Challenge
Hydrogen is playing an increasingly important role across industries, from clean energy and mobility to chemical processing and advanced manufacturing. At the same time, hydrogen poses a well-known risk to structural metals. Through mechanisms such as hydrogen embrittlement, delayed fracture, and stress corrosion cracking, even small amounts of hydrogen can severely reduce material performance and reliability.
At the core of these degradation processes is a fundamental but unresolved challenge: accurately predicting how hydrogen moves through real engineering materials. In practice, hydrogen does not diffuse through an ideal crystal. Instead, it encounters grains, grain boundaries, and other microstructural features that can either slow transport by trapping hydrogen or accelerate it by acting as preferential pathways. Capturing this competing behavior has proven difficult using conventional modeling approaches.
A Multiscale Problem
Many engineering-scale models of hydrogen diffusion build on extensions of Fick’s law and well-established trapping formulations. These approaches are widely used because they are practical, computationally efficient, and effective for a broad range of engineering problems. However, as materials challenges increasingly demand higher fidelity and microstructure sensitivity, the limitations of purely phenomenological descriptions become more apparent. Parameters such as trap densities and exchange rates are often difficult to measure directly and may not transfer robustly across different materials, microstructures, or loading conditions.
At the other end, atomistic-based approaches provide detailed insight into hydrogen-defect interactions and local migration mechanisms, but they struggle to reach the length and time scales required to directly inform engineering-scale models, making the transfer of atomistic information to continuum descriptions non‑trivial.
Published Synopsys Research Linking Atoms to Engineering Models
Researchers at Synopsys have recently published new results that address this challenge by introducing a multiscale modeling framework for hydrogen diffusion in polycrystalline metals. The work directly connects atomic-scale hydrogen migration energetics with continuum-scale transport predictions, providing a physically grounded alternative to traditional trapping-based approaches.
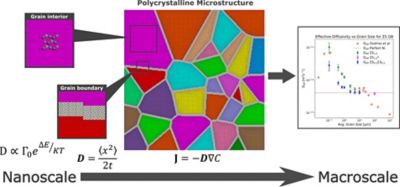
An overview of how hydrogen diffusion is modeled across a polycrystalline material. Atomistic simulations provide diffusion behavior inside grains and along grain boundaries, which is then used to predict effective, large-scale diffusivity as a function of grain size.
At the smallest scale, first-principles calculations are used to quantify hydrogen migration barriers in both crystal interiors and representative grain boundaries. Rather than treating interfaces as abstract traps, their structure and energetics are explicitly considered. This atomistic information is then transferred upward using a mesoscopic step to define effective transport properties that can be used in engineering-scale simulations.
At the continuum level, polycrystalline microstructures are modeled explicitly. Grain interiors are assigned bulk transport properties while grain boundaries are represented as thin regions with anisotropic diffusivity. In this way, trapping and fast-path effects emerge naturally from the spatial variation in material properties rather than being imposed through constitutive terms.
Predicting Effective Diffusivity Across Microstructures
In nickel, highly ordered “special” grain boundaries are shown to behave as diffusion barriers with quantitatively higher migration barriers for hydrogen crossing the interface. In contrast, more disordered grain boundaries exhibit lower effective migration barriers and support markedly higher in‑plane hydrogen diffusivity. The framework computes these differences explicitly, resolving orders‑of‑magnitude variations between transport parallel and perpendicular to the boundary.
A key outcome of the published study is not merely confirming that grain boundaries affect hydrogen diffusion, as this has long been understood, but demonstrating that their impact can now be quantified in a predictive manner. Grain boundary effects are expressed as numerical, direction‑dependent diffusivities derived directly from atomistic energetics rather than inferred qualitatively or fitted phenomenologically.
Using synthetic polycrystalline microstructures, the Ansys, part of Synopsys, framework reproduces classic hydrogen permeation tests in silico. By applying a concentration gradient across a representative volume element and monitoring the resulting flux, an effective macroscopic diffusivity can be extracted.
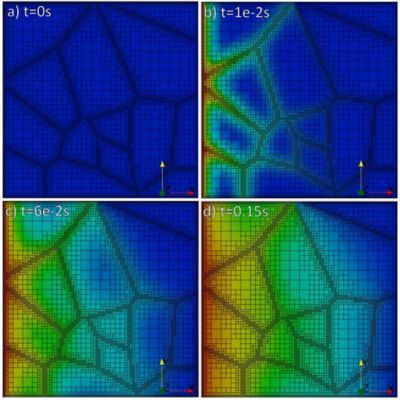
Hydrogen concentration snapshots for a material with 1 μm grains. The images show that hydrogen moves faster along the grain boundaries.
The simulations reveal clear, intuitive trends. As grain size decreases, grain boundaries occupy a larger fraction of the material and increasingly control hydrogen transport. Microstructures dominated by fast-diffusing boundaries exhibit higher effective diffusivity while coarse-grained materials approach bulk crystal behavior.
Equally important, microstructures containing a mixture of fast and slow boundaries show intermediate transport behavior. Even partially replacing fast pathways with slower interfaces can significantly reduce global hydrogen mobility. This sensitivity highlights why grain boundary engineering is an effective strategy for improving resistance to hydrogen-related degradation.
Importantly, these predictions align well with experimental trends across a wide range of grain sizes. Achieving this level of agreement without relying on fitted trapping parameters demonstrates the strength of a physics-based multiscale approach.
Looking Ahead
Beyond matching experiments, the framework provides actionable insight. Because transport properties are directly tied to grain boundary structure, the model helps explain why certain microstructures perform better in hydrogen-rich environments.
While the published study focuses on hydrogen diffusion in nickel, the methodology is broadly applicable. The same multiscale strategy can be extended to other alloys and microstructural features — for example, precipitates — and later coupled with mechanical loading to study stress-assisted hydrogen transport.
For wider adoption, the workflow must be easy to use. Today, the continuum portion is already highly automated and repeatable using PyAnsys libraries, but the atomistic input remains the most complex step (selecting structures, preparing calculations, and curating energetics). One practical option to simplify this stage is to employ machine-learning interatomic potentials (MLIPs) as a possible push‑button source of atomistic diffusivities.
For engineers and materials scientists, the message is clear. Predictive modeling of hydrogen transport is possible. By connecting atomic‑scale physics to engineering-scale simulations, Ansys researchers are enabling microstructure‑informed strategies to design safer, more reliable materials for hydrogen‑critical applications.
Learn More



The Advantage Blog
The Ansys Advantage blog, featuring contributions from Ansys and other technology experts, keeps you updated on how Ansys simulation is powering innovation that drives human advancement.